Physical Address
304 North Cardinal St.
Dorchester Center, MA 02124
Physical Address
304 North Cardinal St.
Dorchester Center, MA 02124

According to 21 CFR Part 3.2, a combination product is a product comprised of two or more regulated components (drug/biologics/device) that are physically or chemically combined or mixed. any combination of a drug & a device or biologics into a single, integrated system. These products are designed to achieve a specific therapeutic outcome, and their components are often combined to improve the overall effectiveness, safety, or convenience of the treatment. They are either combined as single entity items or co-packaged.
Types of Combination Products:
Drug and Device: A product that combines a drug and a medical device They can be co-packaged or as a single entity. For example:
Biological Product and Device: A biological product combined with a medical device. For example:
Drug and Biological Product: A combination of a drug and a biologic. For example:

Combination products are regulated by government agencies (like the U.S. FDA, or the European Medicines Agency (EMA)) and typically require oversight in multiple areas depending on the components involved (e.g., drug, device, biologic). The regulation ensures that all components of the product are safe, effective, and appropriately manufactured for their intended use.
Generally, one investigational application is sufficient for a combination product, which should include all the information on the entire combination product. For example, If it is a drug-device combination product, the application should include the details of the drug that would be submitted in an IND application (Investigational for New Drug) and the details of the device that would be submitted in an IDE (Investigational Device Exemption) application.
The office of Combination Products (OCP) is the focal point for combination product issues and for medical product classification and assignment issues for agency staff and industry. OCP also develops guidance and regulations to clarify the regulation of combination products. They can be reached at combination@fda.gov.
OCP does not review marketing applications for combination products and assigns the lead Center (CBER, CDER or CDRH) that will have primary jurisdiction and responsibility for the premarket review and regulation of a combination.
During the development of combination products, the scientific and technical challenges of the individual components and the whole combination products needs to be considered. Since drugs/biologics and devices are regulated differently, their interactions may not always be immediately clear. Furthermore, due to the complexity and innovation involved, there’s no single development approach that suits all combination products.
Existing guidelines by FDA (found here) or 21 CFR Part 4 regulations are useful for understanding the issues related to the individual parts of a combination product, but they often need to be adjusted to address the unique nature of these products. For example, preclinical guidance for drugs/biologics differs from that for devices, so a combined approach is often needed to address the development needs of both the individual parts and the entire product. The FDA advises developers to consider all scientific and technical aspects and propose a development strategy that avoids unnecessary duplication of studies.
The specific development considerations depend on the type of combination product. We will focus on combination products for biologics, including vials, pre-filled syringes, autoinjectors, and wearables.
Design and development of combination products must follow design control process according to 21 CFR Part 4 regulations and ISO 13485 standard for medical devices.
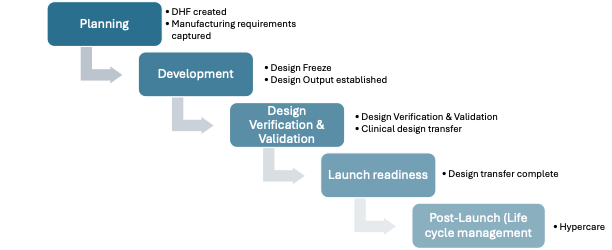
During planning, Design History File (DHF) is generated and manufacturing requirements are captured. Then, user needs are identified and following the design and development, the design outputs are established. Verification assesses if design outputs meet the design inputs and Validation activities evaluates if design outputs meet user needs.
Design transfer to manufacturing and production site is then completed to be ready for launch.
Post-Launch, life cycle management activities to monitor complaints and implement required changes and improvements will take place.
Combination Products need to follow the design control for medical devices according to ISO 13485.
First, user needs to be defined that needs to consider all the key interactions between the user and the device. Then, user needs will be translated into design inputs focusing on the device’s key features and properties. Following the design and development process, design output is generated. The next step is to perform analytics to make sure the output device meets the requirements. Verification tests makes sure that design outputs meet the design input and Validation tests makes sure that design output meets user inputs.
The whole design process needs to be accompanied by risk management procedures according to ISO 14971. Risk management activity identifies the key features that needs more focus and attention to pass the requirements.
We will describe specific examples for each of the design control steps and risk managements for the key combination products for injectable biologics in here.

We are expert in combination products for injectable biologics including prefilled syringes, auto injectors, and wearables. Explore our learning documents and templates.